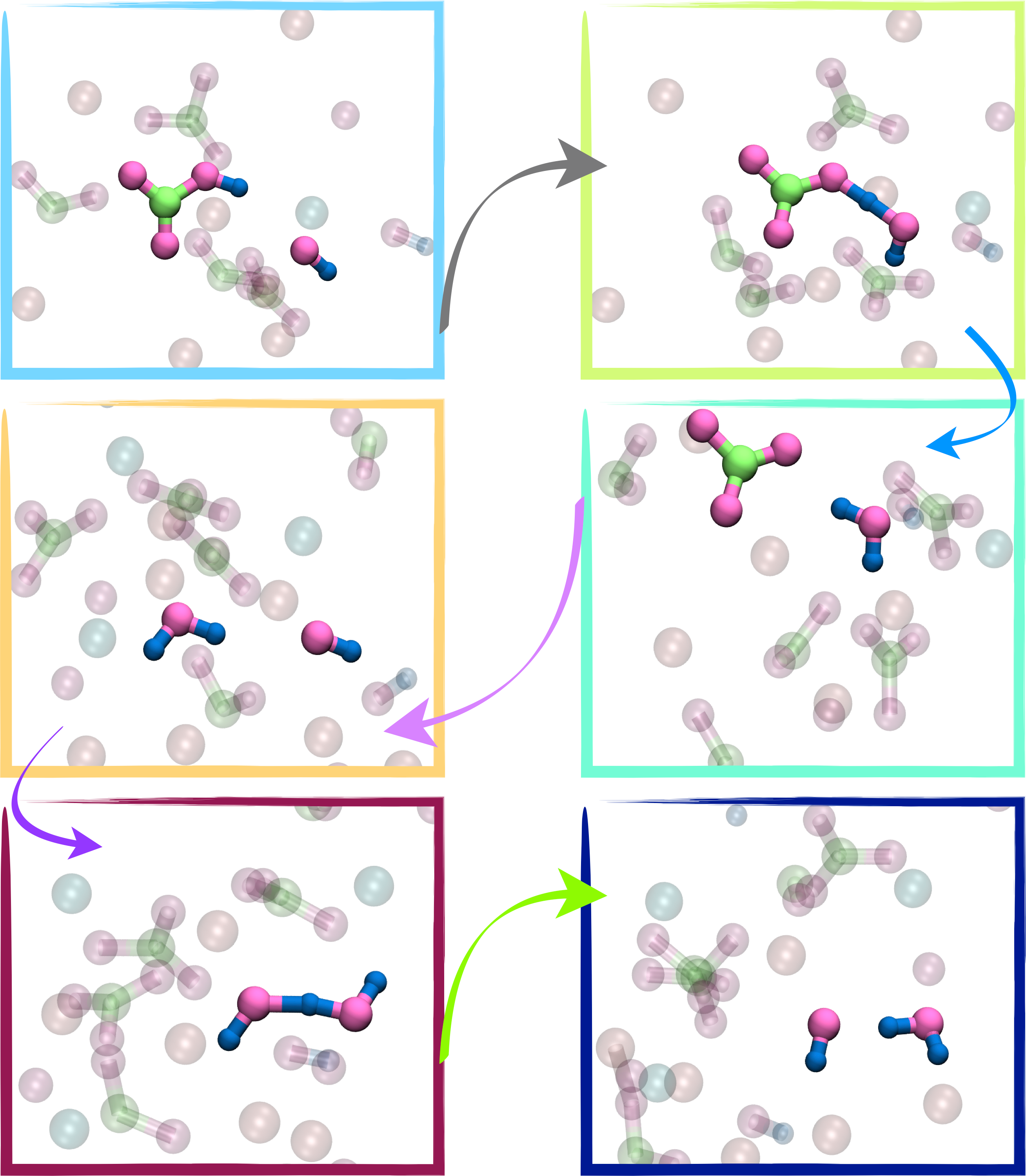
Predictive Modeling of Charge Transport in Organic Semiconductors
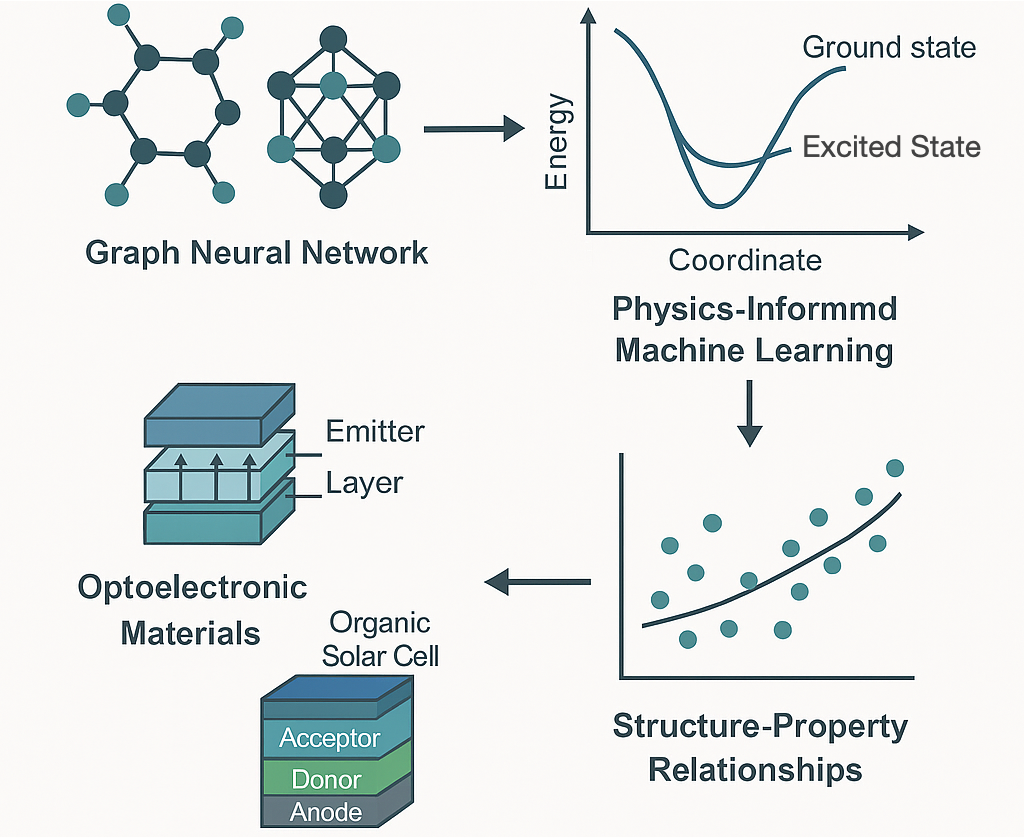
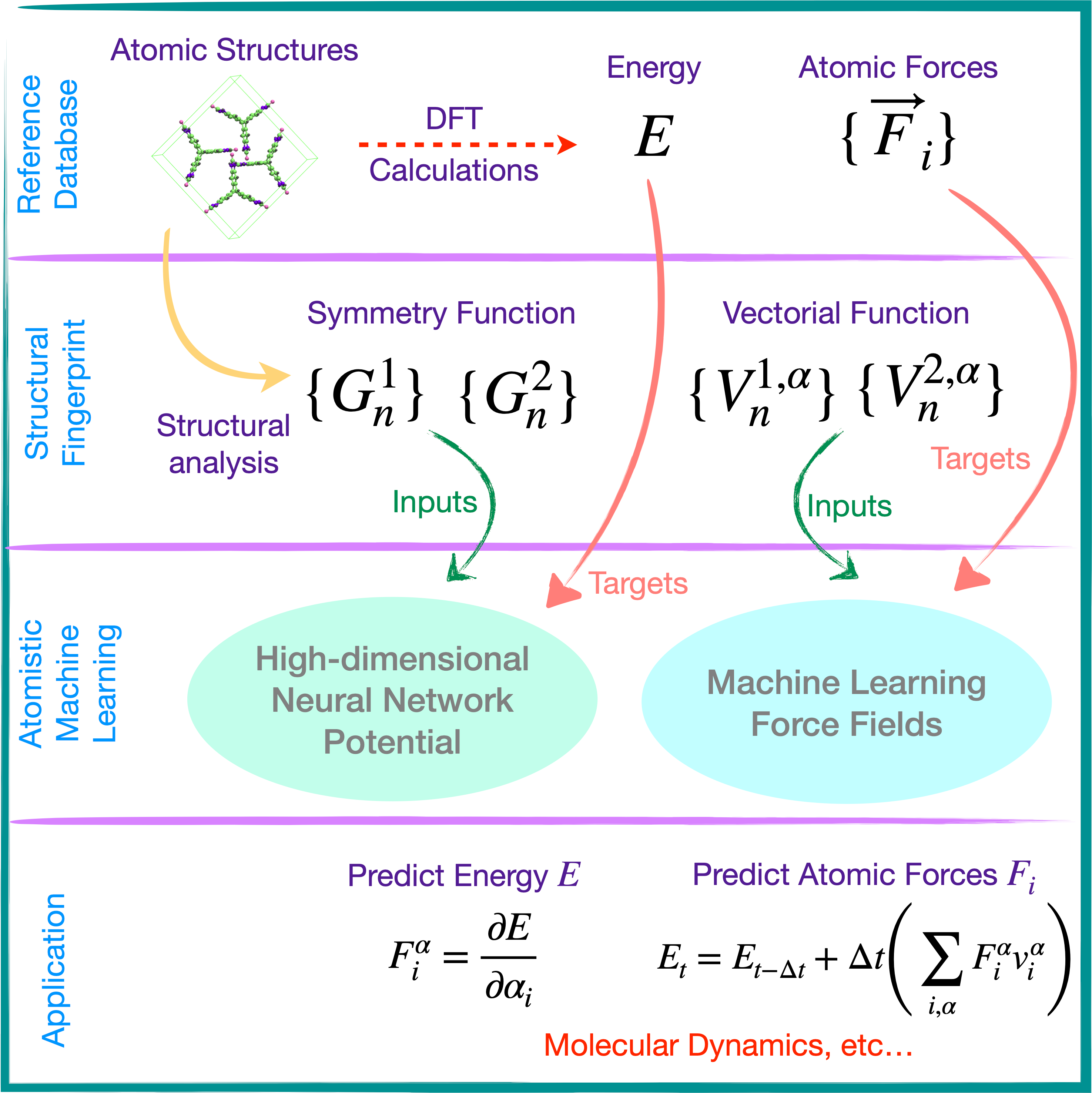
Organic semiconductors are exotic, carbon-based optoelectronic materials that are utilized in a broad spectrum of optoelectronic applications, stretching from organic light-emitting diodes to perovskite solar cells. They exhibit unique solid-state physical properties owing to the soft, van der Waals bonding between individual molecules. The presence of strong coupling between the electronic and structural dynamics gives rise to unique and fascinating phenomena in these molecular solids, such as a transient localization of the electronic states, but is also a performance-limiting factor for their charge transport and optoelectronic properties, such as carrier mobilities or excited-state lifetimes. Using a multiscale computational workflow, we aim to explore bulk charge transport parameters in organic semiconductors and procure molecule-specific quantitative predictions which would empower the prescreening of a diverse set of materials for the design of highly efficient optoelectronic devices.